Maladie de Wilson : accumulation de cuivre et thérapie de chélation
 janv., 19 2026
janv., 19 2026
Qu’est-ce que la maladie de Wilson ?
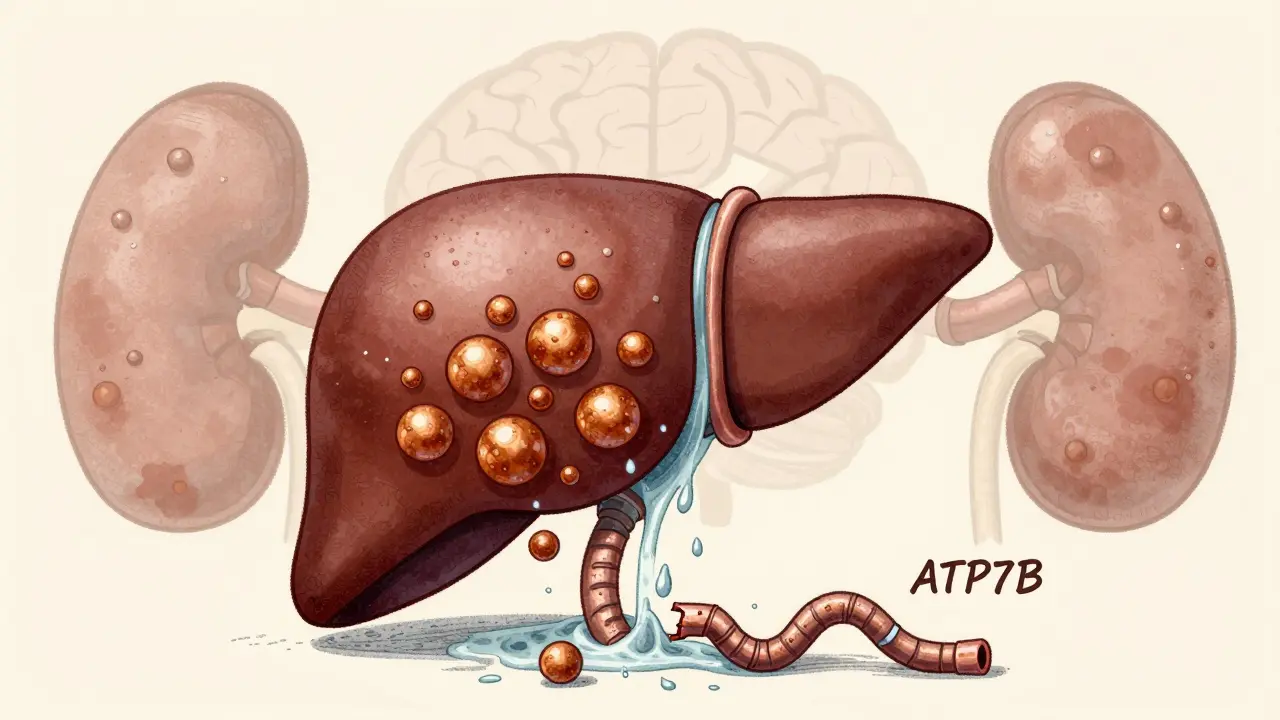
La maladie de Wilson est un trouble génétique rare qui empêche le corps d’éliminer correctement le cuivre. Au lieu de sortir naturellement par la bile, le cuivre s’accumule dans le foie, puis dans le cerveau, les reins et les yeux. Sans traitement, cette accumulation est mortelle. Mais avec un diagnostic précoce et une thérapie adaptée, les patients peuvent vivre aussi longtemps que n’importe qui d’autre.
Ce n’est pas une maladie du cuivre en excès dans l’alimentation. Même avec une alimentation normale, les personnes atteintes ne peuvent pas le traiter. Le problème vient d’une mutation dans le gène ATP7B, situé sur le chromosome 13. Ce gène produit une protéine qui transporte le cuivre dans les cellules du foie. Quand elle est défectueuse, le cuivre s’accumule comme une eau qui ne s’écoule pas.
Comment le cuivre s’accumule-t-il dans le corps ?
Chez une personne en bonne santé, le cuivre entre par l’intestin, puis est acheminé au foie. Là, la protéine ATP7B le lie à la ceruloplasmine - une protéine qui transporte 95 % du cuivre dans le sang - et le libère dans la bile pour qu’il soit éliminé. Chez les personnes atteintes de la maladie de Wilson, cette machine s’arrête. La ceruloplasmine ne se forme pas correctement, et le cuivre ne sort plus par la bile.
Les premières années, le foie capte le cuivre avec des protéines de stockage appelées métallothionéines. Mais quand ces réserves sont pleines, le cuivre commence à fuir dans le sang sous forme libre. C’est ce cuivre libre qui circule et s’accumule dans le cerveau, surtout dans les noyaux gris centraux : le putamen et le globus pallidus. C’est là que les symptômes neurologiques apparaissent.
Un signe presque pathognomonique est l’anneau de Kayser-Fleischer : une décoloration brunâtre autour de la cornée, visible avec un ophtalmoscope. Présent chez 95 % des patients avec des symptômes neurologiques, il est un indicateur fiable - sauf chez les jeunes enfants, où il est souvent absent.
Les symptômes : foie ou cerveau, ou les deux ?
La maladie de Wilson ne se présente pas toujours de la même façon. Certains patients développent d’abord des problèmes hépatiques : fatigue, jaunisse, douleurs abdominales, ou des taux élevés d’enzymes hépatiques. D’autres, surtout adolescents et jeunes adultes, montrent des signes neurologiques : tremblements, raideur musculaire, difficultés à parler ou à avaler, changements d’humeur, ou même des crises psychiatriques.
Les symptômes apparaissent généralement entre 5 et 35 ans. Mais le diagnostic est souvent retardé - en moyenne de 2,7 ans - parce que les médecins pensent d’abord à des maladies plus courantes comme l’hépatite auto-immune. La clé ? Une ceruloplasmine basse (< 20 mg/dL) associée à une excrétion urinaire de cuivre élevée (> 100 μg/24 h). Chez les formes neurologiques, cette valeur peut descendre à 70 μg/24 h, ce qui complique le diagnostic.
Comment diagnostique-t-on la maladie de Wilson ?
Le diagnostic repose sur plusieurs éléments combinés. Il n’y a pas un seul test qui suffise. Les médecins utilisent un système de points :
- Ceruloplasmine sérique basse : 2 points
- Excrétion urinaire de cuivre > 100 μg/24 h : 2 points
- Anneau de Kayser-Fleischer : 2 points
- Concentration hépatique en cuivre > 250 μg/g de tissu sec : 2 points
- Mutation dans le gène ATP7B : 4 points
Un total de 4 points ou plus confirme le diagnostic. Depuis 2023, la présence d’une mutation ATP7B est considérée comme un critère définitif - même sans symptômes. Cela permet de dépister les membres de la famille, puisque la maladie est héréditaire et récessive : les deux parents doivent être porteurs pour qu’un enfant soit touché.
Le test de cuivre urinaire sur 24 heures est crucial. Il montre que le corps tente de se débarrasser du cuivre - mais en vain - ce qui le distingue des autres maladies du foie. Dans l’hépatite auto-immune, le cuivre urinaire est normal. Dans la maladie de Wilson, il est toujours élevé.
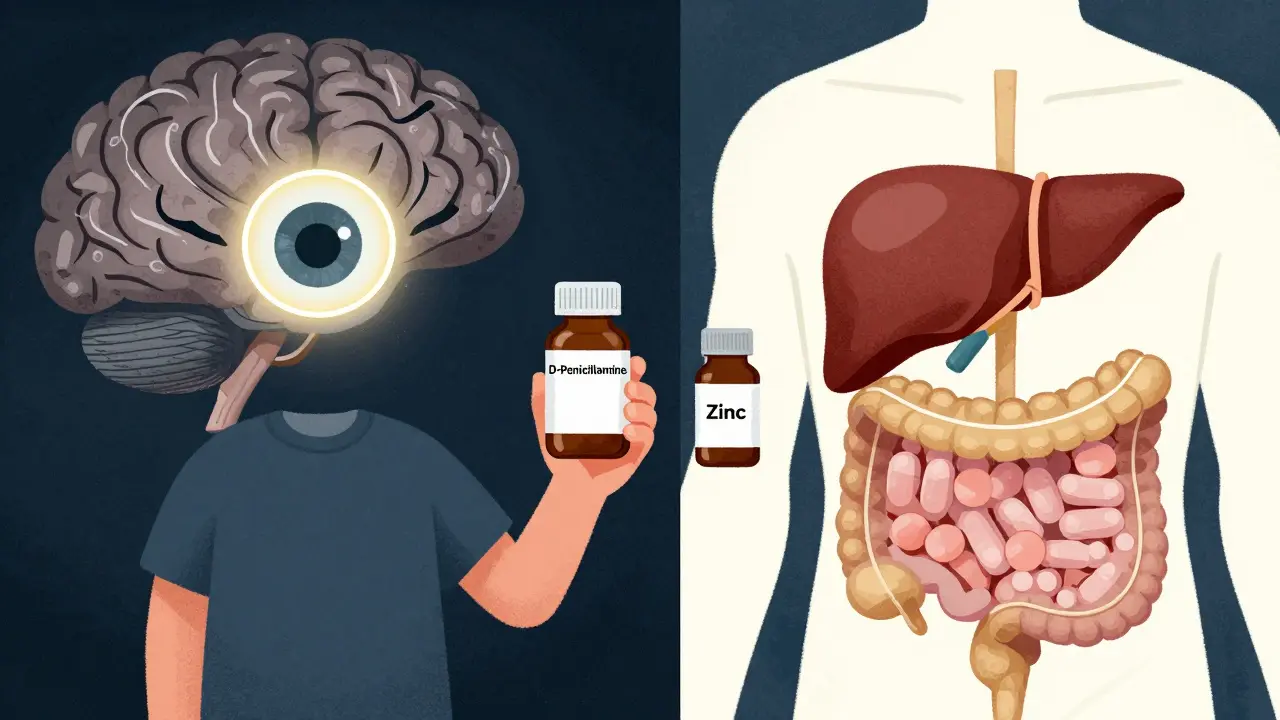
Chélation : le cœur du traitement
Le traitement repose sur deux principes : éliminer l’excès de cuivre, puis empêcher son retour. La première ligne est la chélation - l’usage de molécules qui se lient au cuivre pour le faire sortir par les urines.
D-pénicillamine est le traitement le plus utilisé depuis les années 1950. Elle est bon marché (environ 300 €/mois) et efficace. Mais elle cause des effets secondaires chez 74 % des patients : nausées, goût métallique, réactions cutanées, ou même un syndrome pseudo-lupus. Et dans 20 à 50 % des cas, elle aggrave temporairement les symptômes neurologiques au début du traitement.
Trientine est une alternative plus douce. Moins de réactions allergiques, moins de dégradation neurologique initiale. Mais elle coûte près de 1 850 €/mois - plus de six fois le prix de la pénicillamine. Elle est souvent réservée aux patients qui ne tolèrent pas la première.
Les deux médicaments doivent être pris à jeun, au moins une heure avant les repas. Manger en même temps réduit leur efficacité. C’est un défi pour les patients : devoir planifier leur journée autour de leurs comprimés.
Le zinc : le gardien du cuivre
Après la phase de chélation, le traitement passe en mode maintenance. Le zinc est alors le meilleur allié. Il ne retire pas le cuivre déjà présent, mais il bloque son absorption dans l’intestin.
Comment ? Le zinc stimule la production de métallothionéine dans les cellules intestinales. Cette protéine piège le cuivre de l’alimentation, et l’empêche de passer dans le sang. Il est pris sous forme d’acétate de zinc, trois fois par jour, 30 mg par prise.
Le zinc est idéal pour la vie : sans effets secondaires majeurs, peu coûteux (environ 450 €/mois), et très efficace. Une étude montre qu’il empêche la récidive neurologique dans 92 % des cas, à condition que le cuivre libre dans le sang reste sous 10 μg/dL.
La plupart des patients passent de la pénicillamine ou la trientine au zinc après 6 à 12 mois. C’est une transition cruciale. Beaucoup de patients qui ont eu des effets secondaires graves avec la chélation retrouvent une qualité de vie normale avec le zinc.
Les nouveaux traitements en vue
La recherche avance. En 2023, un nouveau composé appelé CLN-1357 a montré une réduction de 82 % du cuivre libre en seulement 12 semaines, sans aggraver les symptômes neurologiques. Il est encore en phase d’essai, mais il pourrait remplacer les chélateurs traditionnels.
Un autre médicament, le WTX101 (bis-choline tetrathiomolybdate), a reçu le statut de « thérapie révolutionnaire » de la FDA en janvier 2023. Dans un essai, il a prévenu la détérioration neurologique chez 91 % des patients - contre 72 % avec la trientine.
En Europe, le Decuprate® (tetrathiomolybdate de choline) est déjà approuvé pour les formes neurologiques. Il traverse mieux la barrière hémato-encéphalique, ce qui le rend plus efficace pour protéger le cerveau.
À l’horizon, la thérapie génique. Une première expérience chez six patients, avec un virus modifié pour délivrer une version saine du gène ATP7B, a montré une sécurité initiale. Ce n’est pas encore un traitement, mais c’est la première lueur d’une guérison possible.

Les défis du quotidien
La maladie de Wilson ne se soigne pas seulement avec des pilules. Elle demande un changement de vie.
Les patients doivent suivre un régime pauvre en cuivre : moins de 1 mg par jour. Cela signifie éviter les foies, les fruits de mer, les noix, le chocolat, les champignons, les céréales complètes, et même l’eau du robinet si elle passe par des tuyaux en cuivre.
90 % des patients déclarent que c’est difficile. Les repas en famille, les voyages, les restaurants deviennent des épreuves. Certains se tournent vers des diététiciens spécialisés. D’autres se retrouvent avec des carences en fer ou en zinc - parce qu’ils ont supprimé trop d’aliments.
Et puis il y a la question de l’observance. Un patient sur trois oublie au moins une prise par semaine. Les comprimés à jeun, les effets secondaires, la complexité du traitement - tout cela pèse. Les patients qui restent fidèles à leur traitement vivent normalement. Ceux qui l’interrompent risquent une dégradation rapide du foie ou du cerveau.
Prognostic et espoir
Il y a trente ans, la maladie de Wilson était une sentence de mort. Aujourd’hui, elle est une maladie chronique, bien gérée. Avec un diagnostic précoce, un traitement adapté et un suivi rigoureux, les patients atteints peuvent espérer une espérance de vie normale.
Les avancées récentes - nouveaux chélateurs, thérapie génique, diagnostics plus précis - transforment l’avenir. Le vrai défi n’est plus de guérir, mais de dépister plus tôt. Dans les pays à faibles ressources, le diagnostic prend encore cinq ans. Là-bas, la maladie reste fatale.
Partout ailleurs, la clé est la vigilance. Si vous avez des antécédents familiaux, si vos enfants ont des tremblements inexpliqués ou des anomalies hépatiques, demandez un test de cuivre et de ceruloplasmine. Un simple bilan sanguin peut sauver une vie.
Que faire après le diagnostic ?
Une fois le diagnostic posé, trois étapes sont essentielles :
- Commencer immédiatement la chélation (pénicillamine ou trientine) si les symptômes sont présents.
- Prévoir un suivi régulier : cuivre urinaire tous les 6 mois, cuivre libre sérique tous les 3 mois, fonctions hépatiques trimestrielles.
- Passer au zinc dès que possible, pour la maintenance à long terme.
Évitez les compléments en cuivre. Vérifiez les multivitamines. Parlez à votre médecin avant de prendre tout nouveau médicament - certains peuvent interférer avec la chélation.
Et n’oubliez pas : vous n’êtes pas seul. Des associations comme la Wilson Disease Foundation offrent des guides, des forums, et des réseaux de soutien. La maladie est rare, mais les personnes qui la vivent sont nombreuses - et elles partagent des expériences précieuses.
Nathalie Silva-Sosa
janvier 21, 2026 AT 06:03Diane Fournier
janvier 21, 2026 AT 17:08christophe gayraud
janvier 23, 2026 AT 02:31Louis Stephenson
janvier 24, 2026 AT 23:34Seydou Boubacar Youssouf
janvier 26, 2026 AT 13:18Henri Jõesalu
janvier 26, 2026 AT 17:50Nathalie Tofte
janvier 27, 2026 AT 14:08Jean-marc DENIS
janvier 28, 2026 AT 07:21Pastor Kasi Ernstein
janvier 29, 2026 AT 21:32Andre Esin
janvier 30, 2026 AT 10:56